Article Text

Abstract
Background Dementia incidence is increasing across the globe and currently there are no disease-modifying pharmaceutical treatments. The Lancet Commission on dementia identified 12 modifiable risk factors which explain 40% of dementia incidence. However, whether these associations are causal in nature is unclear.
Objective To examine the modifiable risk factors for dementia as identified in the Lancet Commission review using Mendelian randomisation (MR) to establish if, based on genetic evidence, these associations with different dementia subtypes are causal in nature.
Methods Publicly available genome-wide association study data were used for 10 risk factors and Alzheimer’s disease (AD), frontotemporal dementia and dementia with Lewy bodies. Two-sample MR using the inverse varianceweighted method was conducted to test for causal relationships. Weighted median MR and MR-Egger were used to test for pleiotropic effects.
Results Genetic proxied risk for higher levels of smoking (OR: 0.80 (95% CI: 0.69; 0.92), p=0.002), obesity (OR: 0.87 (95% CI: 0.82; 0.92), p<0.001) and blood pressure (OR: 0.90 (95% CI: 0.82; 0.99), p=0.035) appeared to be protective against the risk of AD. Post hoc analyses indicated these associations had pleiotropic effects with the risk of coronary artery disease. Genetic proxied risk of educational attainment was found to be inconsistently associated with the risk of AD.
Conclusions and implications Post hoc analysis indicated that the apparent protective effects of smoking, obesity and blood pressure were a result of survivor bias. The findings from this study did not support those presented by the Lancet Commission. Evidence from causal inference studies should be considered alongside evidence from epidemiological studies and incorporated into reviews of the literature.
- Adult psychiatry
- PSYCHIATRY
Data availability statement
Data are available in a public, open access repository.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
Twelve modifiable risk factors contribute to 40% of the incidence of dementia. When causal inference methods are used to assess these associations, the results are unclear. This may be due to a number of reasons including the inconsistent way genetic instruments are selected and included in these types of analyses.
WHAT THIS STUDY ADDS
This study provides a comprehensive analysis of 10 of these risk factors and 3 different dementia outcomes. A consistent methodological approach was used, and the most recent genome-wide association studies for dementia were used. We did not find robust evidence of causal links between the 10 risk factors and any dementia outcomes.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
The most salient implication of this research is the need to incorporate results from studies using a wide variety of methodological approaches into reviews of the literature. Currently, reviews of the literature in this field are very reliant on studies using traditional epidemiological approaches. However, this study shows that findings from those types of studies are not necessarily backed up by studies using causal inference techniques.
Introduction
Dementia incidence and prevalence rates are steadily increasing across the globe as lifespans increase and the global population ages.1 The quest for pharmaceutical interventions that prevent or modify dementia has thus far proved unsuccessful. Therefore, the spotlight of attention has fallen on lifestyle risk factors, the modification of which may have a role in delaying or preventing dementia. To this end, the Lancet Commission report on Dementia prevention, intervention and care 2 3 has provided the most comprehensively reviewed evidence for potentially modifiable risk factors for dementia. The report highlighted the importance of nine risk factors which were categorised as falling under early-life, mid-life and late-life.3 Less education was identified as an early-life risk factor (<45 years); hearing loss, hypertension and obesity as mid-life risk factors (age 45–65 years) and smoking, depression, physical inactivity, low social contact and diabetes as late-life risk factors (age >65 years). This was further extended to 12 by including the mid-life risk factor of traumatic brain injury and excessive alcohol consumption and the later-life risk factors of air pollution.2 A population attributable fraction (PAF) for each risk factor was calculated, and the authors report that together these 12 risk factors have a combined PAF of approximately 40%. This can be interpreted as: if a causal relationship between the risk factors and dementia exists, then elimination of these risk factors would result in a 40% reduction in the number of dementia cases. The report used the most robust information available, pooling data from epidemiological studies and randomised controlled studies (RCTs). However, it was limited due to assumptions underlying the PAF calculation and the use of all-cause dementia as the outcome.
A key assumption in the PAF calculation and interpretation is that the relationship between the risk factor and outcome is causal. However, although a mixture of study designs examining risk factors was included in the Lancet Commission review, the bulk of the evidence comes from epidemiological studies. A limitation to reporting PAF statistics based on epidemiological studies is that causal relationships cannot be established in these types of observational studies. One reason for this is that observed associations can arise as an artefact of unmeasured confounders. Another reason is that reverse causation cannot be ruled out and this may be particularly problematic when the disease in question has a long preclinical period as is the case with dementia. Therefore, one of the key messages of the Lancet commission report (modifying the 12 lifestyle risk factors may prevent or delay up to 40% of dementias in the population) is based on the assumption that the results of the studies included in the report have provided evidence to support causality.
The second limitation is the use of all-cause dementia as an outcome. Although there are benefits to using all-cause dementia as a generic outcome in terms of power and maximising the number of included studies, a limitation is that each subtype of dementia is a different disorder with differing underlying pathologies. As such it seems likely that each subtype of dementia may be differentially affected by a particular risk factor. For example, evidence from meta-analyses suggests that obesity may pose a greater risk for the development of Alzheimer’s disease (AD) than for vascular dementia.4 These potentially differential effects could not be accounted for in the Lancet Commission report.
One way to address the first of these limitations is by using Mendelian randomisation (MR). MR is a statistical technique that combines genetic data with epidemiological data to overcome the limitations of confounders and reverse causation and is considered analogous to the RCT.5 Within the population, some individuals are more at risk of experiencing certain exposures. For example, some people may be at greater risk of obesity by virtue of their genetic make-up. This risk is ‘allocated’ by genetic meiosis and determined at conception and therefore this natural assortment of risk alleles is considered equivalent to the random allocation process of the RCT. Although population stratification and assortative mating may have an impact on human DNA, reverse causality can be largely ruled out because it is unlikely that the exposures individuals experience throughout the course of their lives will have an impact on their genes. Thus, MR studies can be used to estimate the causal effect of the exposure on the outcome given three assumptions are met. These assumptions underpinning MR analysis are the following: the genetic instrument is associated with the exposure phenotype; the genetic instrument is not associated with any confounders; and the genetic instrument exerts its impact on the outcome via the exposure. Given these assumptions are met, MR studies can be useful tools in determining and providing evidence of causal relationships.
To date, evidence coming from MR studies investigating the relationship between risk factors and dementia has been mixed. One potential reason for the mixed findings is the discordant application of instrument selection across studies and subsequent analysis pipelines. Therefore, we aim to conduct a comprehensive MR analysis of as many as possible of the modifiable risk factors for dementia as identified by the Lancet Commission using a standardised rigorous pipeline to allow for the comparability of results. In addition, to address the limitation of using all-cause dementia as a generic outcome we aim to conduct the analysis for three different subtypes of dementia: AD dementia, dementia with Lewy bodies (DLB) and frontotemporal dementia (FTD), for which genome-wide association study (GWAS) data exist. We aim to use the most recent GWAS releases based on the largest sample sizes thus creating the most up-to-date and comprehensive MR analysis on risk factors for dementia.
Method
Study design
GWAS data were available for 10 out of the 12 Lancet Commission risk factors for dementia, and data were not available for traumatic brain injury or pollution. Two-sample Mendelian randomisation analysis was used to investigate potential causal relationships between 10 of the Lancet Commission risk factors for all-cause dementia and 3 different subtypes of dementia: AD (using 4 different GWAS datasets), DLB and FTD. We followed the reporting guidelines provided by the Strengthening the Reporting of Observational Studies in Epidemiology—Mendelian Randomisation.6
Data sources
The current study used publicly available summary statistics and therefore no additional ethics or approvals were required. We searched the GWAS catalogue (https://www.ebi.ac.uk/gwas/) to find the latest GWAS studies for dementia outcomes and the Lancet Commission risk factors for dementia. Publicly available GWAS summary statistics were obtained for each of the 10 risk factors and the 3 dementia-related outcomes (table 1).
GWAS datasets used in the study
Mendelian randomisation analysis
Genetic instruments
For each of the 10 risk factors, single nucleotide polymorphisms (SNPs) were selected based on the genome-wide significance threshold of p <5×10−8. The exposure dataset was merged with the outcome dataset by aligning the effect alleles to the forward strand. The overall F statistic for each exposure was calculated and used as a measure of instrument strength. Only instruments with an F statistic value >10 were used in the analyses to minimise any effects of weak instrument bias. Once the SNPs which met the threshold for genome-wide significance were identified, these SNPs were clumped to remove SNPs in linkage disequilibrium using a threshold of r 2 <0.001 and pruned by selecting the SNP which had the lowest p value. The association between each individual SNP and the dementia outcome was calculated using the Wald ratio,7 and these individual estimates were combined to provide an overall estimate using the inverse variance weighting (IVW) method. To test for potential pleiotropic effects, a sensitivity analysis using MR-Egger and weighted median MR was conducted. Cochran’s Q statistic was used to estimate heterogeneity and as an additional test of horizontal pleiotropy.
As 10 tests were conducted per dementia outcome, a Bonferroni-corrected p value was used and set at p <0.005 to account for multiple comparisons. All estimates were calculated using standardised betas (log OR) and then transformed to the OR. For risk factors measured on a continuous scale, ORs estimated the increased risk per 1 SD increment in the risk factor.
Post hoc analyses were carried out to test a further hypothesis that the effects of obesity, blood pressure and smoking on AD were a result of survivor bias.8 Multivariable MR was conducted to ascertain if pleiotropic effects existed between the three primary risk factors and coronary artery disease (CAD) as a potential pleiotropic pathway, which is consistently reported as the leading cause of mortality globally.9 Summary statistics for CAD were taken from the GWAS as published by Nelson et al (2017).10 Independent SNPs11 for the primary exposure of interest (ie, systolic blood pressure (SBP), body mass index (BMI) or smoking) were selected using the genome-wide significance level of p <5×10−8, clumped to remove SNPs in linkage disequilibrium using a threshold of r 2 <0.001 and pruned by selecting the SNP with lowest p value.
Sensitivity analyses
A sensitivity analysis for each of the 10 risk factors was conducted using 3 additional GWAS datasets for AD. The first of these was a GWAS dataset published by Kunkle et al 12 and the second was a dataset published by Lambert et al.13 The third dataset was the GWAS published by Schwartzentruber et al,14 which combines existing GWAS data on AD with a proxy measure of familial genetic risk.
The main analysis was conducted using data from the largest GWAS on AD and comprised a sample size of 788 989 participants.15 This dataset offered the greatest power to detect causal effects; however, the inclusion of a proxy diagnosis of AD may be vulnerable to disease misclassification and thereby add additional heterogeneity to the results. The Kunkle et al 12 and Lambert et al 13 datasets were used in the sensitivity analysis; although comprising much smaller samples (n=94 437 and n=74 046, respectively), they both used more reliable methods of disease classification. Schwartzentruber et al,14 had a sample size of 408 942 and relied on mostly proxy reports of family history of AD diagnosis was used as an additional dataset to establish the overall replicability of the results.
All analyses were conducted using RStudio (2020) V.1.3.1093 and the ieugwasr and the MendelianRandomization (V.0.5.0)16 R packages. Code was available from: https://github.com/Roop-hub/Examining-the-Lancet-Commission-Risk-Factors-for-Dementia-Using-Mendelian-Randomization.
Results
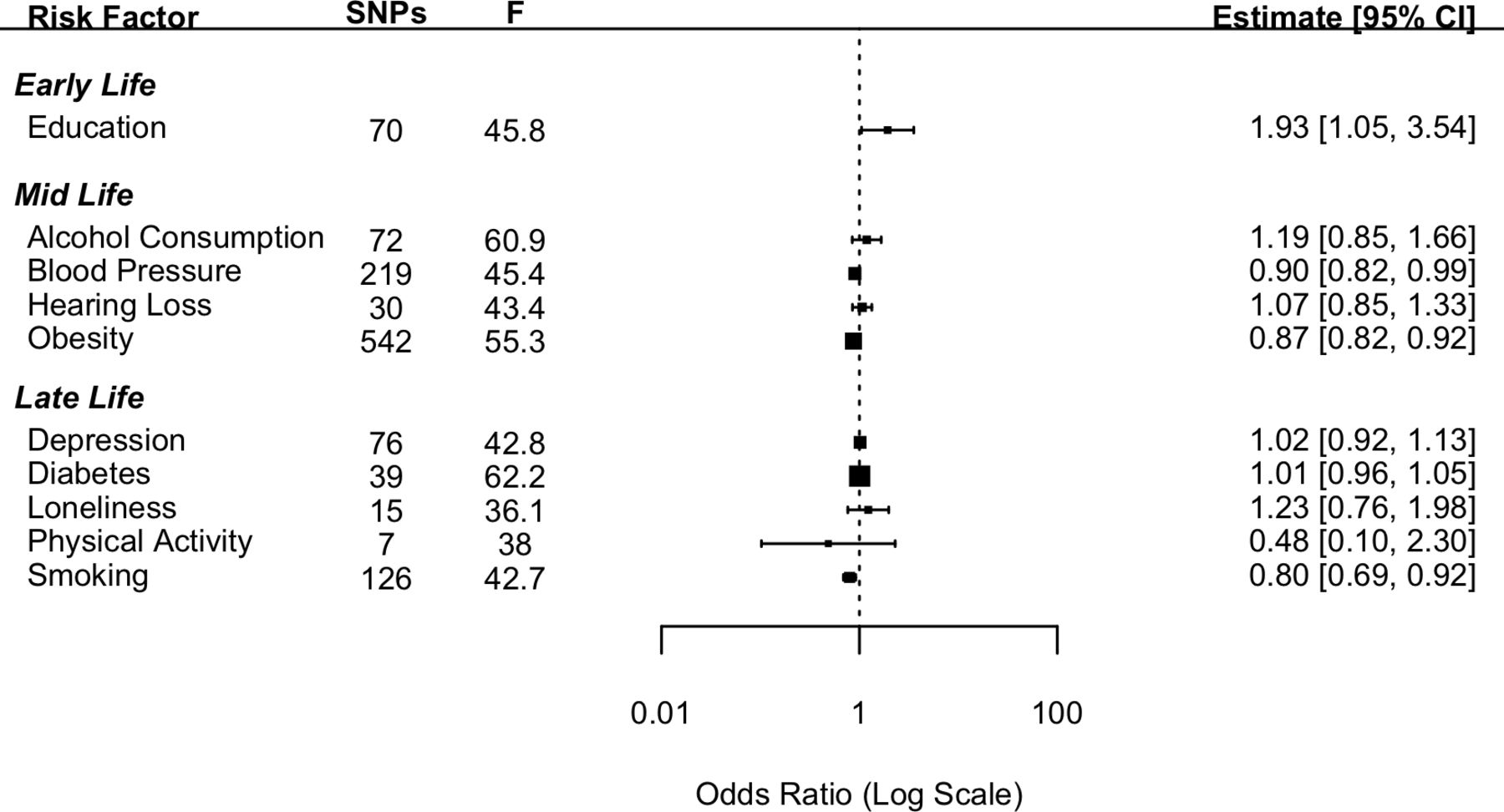
Ten established risk factors for dementia as identified in the Lancet Commission report2 were investigated for their causal effect on three dementia outcomes using two-sample IVW MR. The number of SNPs for each exposure and the test of instrument strength (F statistic) are reported in figures 1–3.

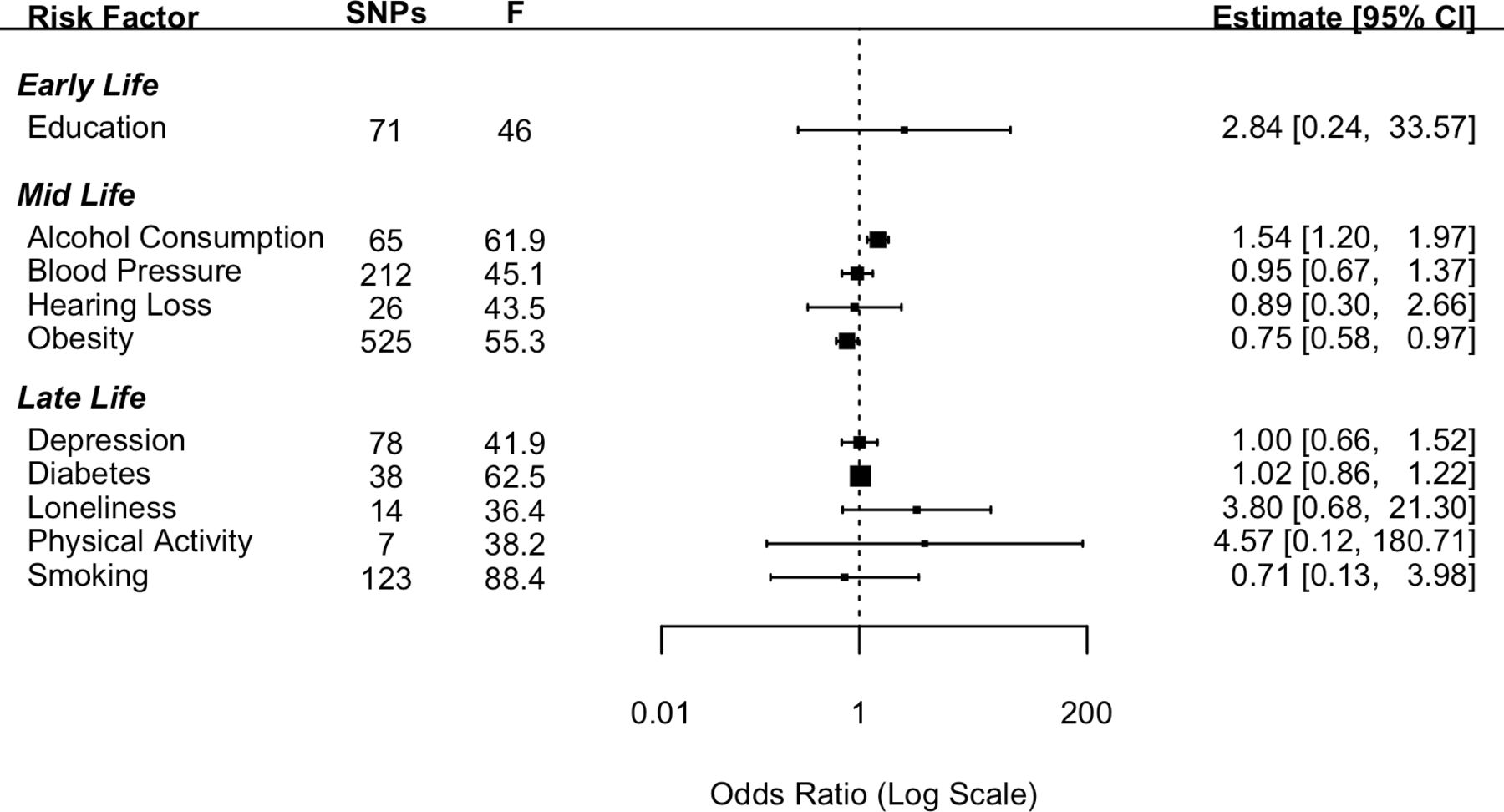
Forest plot showing inverse variance weighted causal effect estimates of modifiable risk factors on Alzheimer’s disease. The number of single nucleotide polymorphisms in each instrumental variable is reported with the corresponding F statistic.

Forest plot showing inverse variance weighted causal effect estimates of modifiable risk factors on dementia with Lewy bodies. The number of single nucleotide polymorphisms in each instrumental variable is reported with the corresponding F statistic.
{kind=link}
{kind=link}
{kind=link}
Forest plot showing inverse variance weighted causal effect estimates of modifiable risk factors on frontotemporal dementia. The number of single nucleotide polymorphisms in each instrumental variable is reported with the corresponding F statistic.
Alzheimer’s disease
Genetically determined higher educational attainment was found to increase the risk of dementia (OR: 1.93 (95% CI: 1.05; 3.54), p=0.03). However, this effect did not meet the criteria for statistical significance after a Bonferroni correction was applied. The mid-life risk factor of genetically determined obesity was found to have a protective effect against the genetic risk of AD with the risk of AD reducing by 13% (OR: 0.87 (95% CI: 0.82; 0.92), p<0.001) with each standard increment in BMI-adjusted waist:hip ratio. The mid-life risk factor of genetically determined lifetime smoking was found to have a protective effect on the risk of AD with a 20% reduction in the risk of AD associated with 1 SD increase in lifetime smoking index (OR: 0.80 (95% CI: 0.69; 0.92), p=0.002). Genetically determined higher SBP was also found to be protective against the genetic risk of AD; however, the estimate did not meet the threshold for Bonferroni-corrected significance (OR: 0.90 (95% CI: 0.82; 0.99), p=0.035).
None of the other risk factors investigated in the current study were found to have a significant effect on the genetic risk of AD (figure 1). Pleiotropy was investigated by conducting MR-Egger analyses. The pattern of results was similar to those attained from the IVW method (online supplemental file 1).
Supplemental material
Sensitivity analyses
For the early-life risk factor of education, contrary to the results from the main analysis the two datasets which measured AD using clinical diagnosis found greater education to be protective against the risk of AD. For the mid-life risk factor of obesity, in keeping with the results from the main analysis, the results from all three additional datasets replicated the protective direction of effect. For the mid-life risk factor of SBP, in keeping with the main analysis all three additional datasets indicated a protective direction of effect. In the case of the late-life risk factor of smoking, the results were mixed.
MVMR analyses
The effects of obesity, smoking and blood pressure were found to be in a direction contrary to the findings reported in the Lancet Commission. That is, the genetic risk of higher obesity, smoking and blood pressure were protective factors against the genetic risk of AD. Given these results, post hoc analyses were conducted using a multivariable MR approach to ascertain if these findings were driven by the pleiotropic effect of CAD. The multivariable MR analyses indicated that the apparent protective effect of all three of these risk factors was driven by the pleiotropic effect of CAD. When CAD was added to the model, the effects of obesity, blood pressure and smoking were attenuated. In the case of blood pressure (OR: 0.95 (95% CI: 0.84; 1.06), p=0.36) and smoking (OR: 0.90 (95% CI: 0.77; 1.06), p=0.20), these effects became non-significant. In the case of obesity (OR: 0.90 (95% CI: 0.84; 0.96), p=0.002) although the effect remained significant, it was greatly attenuated. This suggests that these apparent protective effects on AD are due to the pleiotropic effects of CAD.
Other dementias
Dementia with Lewy bodies
The MR analysis using the 10 exposure risk factors was repeated using DLB as the outcome. Neither educational attainment nor any of the other nine risk factors had a significant effect in relation to DLB (figure 2). Pleiotropy was investigated by conducting MR-Egger analyses. The pattern of results was similar to those attained from the IVW method (online supplemental file 1).
Frontotemporal dementia
The MR analysis using the 10 exposure risk factors was repeated using FTD as the outcome (figure 3). Pleiotropy was investigated by conducting MR-Egger analyses. The pattern of results was similar as those attained from the IVW method (online supplemental file).
Discussion
This study aimed to examine 10 of the 12 risk factors for dementia as identified by the Lancet Commission review2 using MR and a rigorous pipeline analysis to establish if these associations are causal in nature. We used the most recent GWAS data for AD, which is based on the largest sample size. We found apparent protective effects of increased obesity, blood pressure and smoking. Additional post hoc analyses indicated that these apparent protective effects may be due to pleiotropic effects of CAD. There was inconsistency in the direction of effect for educational attainment dependent on the outcome of AD GWAS. None of the other risk factors were found to be significantly associated with the risk of AD. We did not find evidence that any of the 10 risk factors were significant causal risk factors for DLB or FTD.
Contrary to the findings of the Lancet Commission,2 there was evidence that increased obesity, blood pressure and levels of smoking were protective factors in the risk of developing AD. The direction of this effect was replicated using the three additional AD datasets. The results for blood pressure showed a similar pattern with estimates from all four AD datasets indicating a protective direction. In the case of smoking analyses from two14 15 out of the four datasets indicated a protective effect. In a post hoc analysis, we tested the hypothesis that these relationships were a result of survivor bias and driven through the pleiotropic pathway of CAD. Survivor bias has been highlighted as a threat to causal inference in MR studies.8 This is particularly of relevance where the outcome affects an older population as is the case for most dementias. The underlying threat coming from survivor bias in these types of studies is that those individuals who live long enough to develop an age-related disease are the ones who have not died earlier from other morbidities. Thus, what appears to be a link with dementia may be an association with longevity. To test this, we conducted a post hoc multivariable MR and found that the effects of smoking and blood pressure attenuated to a non-significant effect when conditioning on CAD. In the case of obesity although the effect remained significant, it was greatly reduced. This attenuation in effect supports the hypothesis that these results were due to survivor bias. Although we demonstrated survivor bias through the pleiotropic pathway of CAD, it should be noted that other potentially life-limiting diseases such as cancer may also have pleiotropic pathways between the risk factors in question and AD. In addition, alternative explanations for counterintuitive findings should not be discounted. For example, in the case of smoking there is evidence that smoking can be both a risk or a protective factor depending on the dementia type.17 Another factor that should be considered is whether the effects of demographic and indirect effects are adequately controlled for. Growing evidence suggests that GWAS studies from purportedly unrelated individuals do not completely exclude genetic variation due to demographic elements (eg, population stratification and assortative mating).18 This uncontrolled genetic variation can lead to a potential bias in the estimate of the direct effect between genetic variants and the phenotype in question. Within-sibship GWAS analyses more accurately account for demographic effects and should be used in future studies to further explore the associations between risk factors and dementia.
Educational attainment is well established from observational studies as having a protective role in dementia risk. However, our results were inconsistent with opposite effect directions found across different datasets. These inconsistent findings replicate a previous study which found that when proxy information on AD diagnosis is included in the GWAS, as in the Bellenguez et al (2022)15 data, then greater educational attainment is associated with an increased risk of AD.19 One explanation for this finding is that the heterogeneity with the phenotype is greatly increased when a proxy diagnosis is used. Proxy diagnoses rely on individual participants knowing the difference between dementias which may not be reliable. This reliance on lay diagnosis is potentially vulnerable to misclassification thereby increasing the heterogeneity of the SNPs that are associated with the outcome. None of the other risk factors examined were significantly associated with any of the dementia outcomes.
We were not able to completely rule out that the null findings from this study are a consequence of limited power. In additional analyses (online supplemental file 2, online supplemental file 1) we found evidence that genetic risk of higher BMI, SBP and smoking has strong positive causal associations with CAD. These positive associations suggest that if there had been similar effects of these risk factors on AD, we would have been able to detect them. Notwithstanding the limitations of MR studies and that of interpreting null findings, our study did not find evidence to support the risk factors as proposed by the Lancet Commission are causal in nature.
Supplemental material
Supplemental material
Limitations and further research
A strength of the current study was running the analysis for different subtypes of dementia. However, one limitation was that only three were analysed. Thus, we were not able to provide a comprehensive analysis for all common subtypes of dementia. One obvious omission is that of vascular dementia. At the time of completing these analyses, no GWAS studies were available for the genetic risk of vascular dementia. In time there may be GWAS studies for vascular dementia and future similar studies should aim to include this as an outcome. On a related note, with some of the rarer forms of dementia such as posterior cortical atrophy (PCA) although GWAS data do exist for this phenotype, the sample sizes are relatively small. For example, one study only included a sample of 302 PCA cases20 and this was considered by the study authors not sufficiently powered to include in the current analysis. Another limitation of the present study was the inability to stratify the data by APOE-e4 carrier status and sex. APOE-e4 is a risk factor for AD,21 and carrier status may interact with risk factors in a way that produces different outcomes in carriers versus non-carriers.22 For example, APOE-e4 status is associated with slower cognitive decline in women with coronary artery risk factors22 and in APOE-e4 carriers with AD treated for hypertension.23 A further limitation of this study is that the analysis was conducted using datasets of European population samples. Therefore, it is not known if these results are generalisable to other ethnic populations. Currently, the large GWAS datasets required for MR analysis for different ethnicities are not available. Again, with time and more data, these GWAS may increase in sample sizes and diversity of population sample and allow researchers to conduct similar research on rarer forms of dementia and different ethnic groups.
Conclusions
The results of our study found limited genetic evidence for causal links between 10 of the risk factors identified by the Lancet Commission and dementia. There are limitations to results from MR studies, such as that of survivor bias, and therefore the inferences that can be drawn from such studies. However, the results from this study do not align with the findings of the Lancet Commission. Given the limitations to both design types, evidence from causal inference studies should be considered alongside evidence from traditional epidemiological studies and incorporated in reviews of the literature and when applying statistics at a population level.
Data availability statement
Data are available in a public, open access repository.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
VZ and JS are joint senior authors.
Contributors RD, VZ and JS came up with the initial conceptualisation, study design and analysis plan. RD, VZ and RS conducted the data analysis with RD taking the lead and VZ and RS validating the analysis. RD was responsible for writing the manuscript with intellectual input and critical evaluation from all coauthors. JS and VZ provided supervision. RD and JS are the guarantors of the study.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.